Volume of distribution
Note that this chapter relates to the in vivo imaging with radioligands, and does not directly comply with the term volume of distribution as used in pharmacology. In pharmacology, distribution volume of a drug refers to the volume of plasma that would be required to account for the amount of drug in the entire body. Thus, the units of pharmacological volume of distribution are volumes, for example litre (L).
Volume of distribution (VT) of a radioligand is defined as the ratio of the radioligand concentration in tissue target region (CT, kBq·cm-3) to that in plasma (CP, kBq·mL-1) at equilibrium. CP represents the concentration of unchanged radioligand in plasma, that is plasma concentration that is corrected for any radioactive label-carrying metabolites.
Therefore, a VT equal to 15 mL·cm-3 means that the radioligand is being concentrated in tissue by 15:1.
|
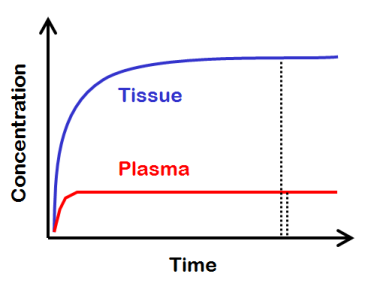
Lets imagine a theoretical situation, where we keep the concentration of radioligand in plasma at a steady level for a very long time. If there is no irreversible uptake of the radiopharmaceutical in the tissue, then, if we wait long enough, we achieve a dynamic equilibrium, where the flux into tissue and out of tissue is the same, and the tissue concentration remains steady. |
|
|
All tissue compartments, where the radiotracer can go in, would then be in equilibrium with all the other tissue compartments, and also with the plasma compartment. Then, the tissue-to-plasma ratio equals the total distribution volume of the radioligand. |
|
Volumes of distribution are applicable to all PET radiopharmaceutical studies, but mainly used in receptor studies. In receptor studies, tissue may contain radioligand that is
- specifically bound to receptors (S),
- nonspecifically bound to other macromolecular components (NS), or
- free in tissue (FT), dissolved in water
CP includes both free and plasma protein bound radioligand. When equation 2 is substituted in equation 1:
, or, if concentrations of free and nonspecifically bound ligand are combined as non-displaceable concentration, CND, then
Thus, the non-displaceable (ND) radioligand uptake is the sum of nonspecific (NS) and free (FT) radioligand concentrations in tissue. This uptake can not be abolished by adding large amounts of non-labelled ("cold") ligand that binds to the same receptor.
| Subscripts abbreviations: | |
|---|---|
| Abbreviation | Description |
| FP, FT | Free radioligand in plasma or tissue |
| ND | Non-displaceable uptake (free + nonspecific) |
| NS | Nonspecific binding |
| P | Plasma free + protein-bound |
| S | Specifically bound radioligand |
| T | Total radioligand in tissue |
Volume of distribution and compartmental model
Volume of distribution is not dependent on compartmental model, although it can also be derived from the differential equations of compartmental models.
Three-tissue compartmental model (parallel compartments)
Two-tissue compartmental model
One-tissue compartmental model
VT can be derived from any compartmental model, as long as there is no irreversible uptake in any of the compartments.
Perfusion
VT is not affected by perfusion, because perfusion is a component of both K1 and k2, and it is therefore cancelled out. This has been confirmed experimentally and in simulations (Holthoff et al., 1991; Logan et al., 1994; Sander et al., 2017). Perfusion does not affect the assessment of receptor occupancy by endogenous ligand either.
Volume of distribution and binding potential
For a receptor-binding ligand, VT is proportional to both receptor density (concentration of available receptors, Bavail) and the binding affinity (KD):
VT is also affected by plasma protein binding and nonspecific binding in tissue.
Therefore, binding potentials are defined: binding potentials are less dependent on these confounding factors, and therefore more directly related to the concentration and affinity of receptors in the tissue.
Plasma or blood?
Most often drug and PET radiopharmaceutical concentrations are measured in plasma or serum and not in whole blood, and simply therefore plasma or serum is also the reference fluid for the volume of distribution. However, whole blood, not plasma or serum, is flowing in the capillaries. Most drugs as well as PET radiopharmaceuticals are bound to plasma proteins and/or partitioned into red blood cells. It is commonly assumed that only free drug or radiopharmaceutical in the plasma water can leave the capillary space. However, if repartitioning of drug or radiopharmaceutical is fast compared with the residence time of blood in the capillaries, then whole blood would be more appropriate reference fluid (Hinderling, 1997).
Body water
The percentage of body weight attributed to water is ∼60%, depending on the amount of adipose tissue. About 2/3 of total body water is intracellular, and the remaining 1/3 is extracellular. About ¾ of extracellular water is in interstitial space, and ¼ in blood plasma.
Macromolecules account for ∼¼ of the cell mass. Structured hydration shell of water surrounds the macromolecules, and most of the intracellular water is interfacial to macromolecules. This "structured water" affects the binding and biochemical reactions.
See also:
- Compartmental model
- Binding potential
- Receptor occupancy (RO)
- Calculation of Logan plot for regional data
- Calculation of Logan plots for images
- Equations for graphical analysis of reversible tracers (Logan plot)
References:
Hinderling PH. Red blood cells: a neglected compartment in pharmacokinetics and pharmacokinetics. Pharmacol Rev. 1997; 49(3): 279-295.
Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN, Holden J, Houle S, Huang SC, Ichise M, Iida H, Ito H, Kimura Y, Koeppe RA, Knudsen GM, Knuuti J, Lammertsma AA, Laruelle M, Logan J, Maguire RP, Mintun MA, Morris ED, Parsey R, Price JC, Slifstein M, Sossi V, Suhara T, Votaw JR, Wong DF, Carson RE. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab. 2007; 27(9): 1533-1539. doi: 10.1038/sj.jcbfm.9600493.
Slifstein M, Laruelle M. Models and methods for derivation of in vivo neuroreceptor parameters with PET and SPECT reversible radiotracers. Nucl Med Biol. 2001; 28(5): 595-608. doi: 10.1016/S0969-8051(01)00214-1.
Tags: Volume of distribution, Binding potential, Specific binding, Non-displaceable uptake, Modeling, Macroparameter, Plasma protein binding, Compartmental model, 1TCM, 2TCM, 3TCM
Updated at: 2019-01-16
Created at: 2014-05-02
Written by: Vesa Oikonen